#S25
Avtor: Ava Boštjančič
Datum izdelave: 2024-05-26
Koda seminarja: S25
##Vhodni podatek Povezava do datoteke z vhodnim podatkom: S25
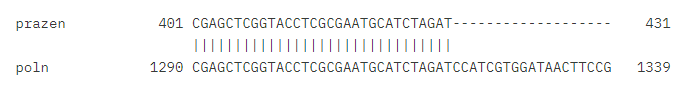
##Rezultati analiz ###Ime in izvorni organizem proteina Vhodni podatek vsebuje nukleotidno zaporedje plazmida pUC57 z vstavljenim neznanim nukleotidnim zaporedjem, ki zapisuje del proteina. Najprej poiščemo sekvenco “praznega” plazmida in nato z lokalno poravnavo (EMBOSS Water) našega in praznega plazmida poiščemo sekvenco in lokacijo vstavljenega zaporedja. Za poravnavo uporabimo prednastavljene parametre. Zaporedji plazmidov se popolnoma prekrivata razen na mestu vstavljenega zaporedja. Začetek in konec vstavljenega zaporedja vidimo na sliki 1 in sliki 2. Iz slik lahko razberemo, da se naše vstavljeno zaporedje nahaja med nukleotidi 1321-2233.
Slika 2:
Nukleotidno zaporedje vstavljenega zaporedja je: CCATCGTGGATAACTTCCGGCATAGAACCCTTATTAAAAGCGATGACTGGAGTTCCACAGGCCATGGCTTCCACCACGCTGAGGCCAAAGGGTTCATGGAAATGAATGGGATGAATAAGAGCCAGGGCATTTCCCATAAGCTGATTGCGCTGGGCAGGATCAGCTACACCATAATAAATGATATTCTCCCCATCGATAAATGGCTCGACCCGGATATTAAAGTAGTCCTGGTCTTGGATTATCCCTGCTATTATGAGCTTAATACCTGACTTTTTTGCAATTTCCACCGCCTCTGCCGTACCTTTATCAGGATGGATCCTGCCAAAATACAACAGGTAATCTCCAGCTTTTTCATGGAAGCTGAACTGCTGAAGGTCGATGCCATGGTAAATGGTTTTTATATAATCCAAATATTCGCTGCGATCCGCGTTGCTTATAGAGACATAATAATTACTATCATTGTATTCCCGATATACAGGTATTATTTGATTGGAGGAAAAACCATGTATGGTAGTTACCACTGGAGTATCTACCAGTCTGGTAAAAACCAGCGGCAAAAAGTCATAATTATTGTGGATAATATCAAATTTATCAGCTTCTTCGAACAAGTGCGCAATGTGCAACGCTTCCCATACTTTAGGGTCAATATCTTGATTTTCTTCATATGGTGTAGGGCAAATAGACGCTAGTTGGGCCCCGGTTATAGAATCTCCGGTAGCGAAGAGCGTTACCTGTAGACCTCTTTTTATCAGTCCTTCAGCCAGCAGGGATACTACTAATTCCCAGGGGCCGTAATTCCGGGGCGGGGTTCTCCAGGCAATAGGTGATAACAATGCTATTTTCATTTACCATGCTCCTCAAAATATTGTCGTAAAGATTTTTTGTAACTCGAAAAATCCAGGTCGATTTTACC
Ko poznamo sekvenco našega vstavljenega nukleotidnega zaporedja, nas zanima prevedeno aminokislinsko zaporedje. Z blastx iščemo podobno aminokislinsko zaporedje po metagenomskih proteinih. Najdemo zaporedje hipotetičnega proteina s 100 % podobnostjo in nizko E-vrednostjo z oznako AZS_017360. Našli smo celotno aminokislinsko zaporedje našega vstavljenega nukleotidnega zaporedja. Naš protein ima anotirano GT4_AviGT4-like domeno z vezavnim mestom za UDP-glukozo in kofaktorjem tetrahidrobiopterinom. Protein je razvrščen v rod bakterij Syntrophomonas.
Celotno aminokislinsko zaporedje našega proteina je: MKIALLSPIAWRTPPRNYGPWELVVSLLAEGLIKRGLQVTLFATGDSITGAQLASICPTPYEENQDIDPKVWEALHIAHLFEEADKFDIIHNNYDFLPLVFTRLVDTPVVTTIHGFSSNQIIPVYREYNDSNYYVSISNADRSEYLDYIKTIYHGIDLQQFSFHEKAGDYLLYFGRIHPDKGTAEAVEIAKKSGIKLIIAGIIQDQDYFNIRVEPFIDGENIIYYGVADPAQRNQLMGNALALIHPIHFHEPFGLSVVEAMACGTPVIAFNKGSMPEVIHDGKSGFLVKDVEQACSVLPEVLSLDRSYCRFWAESRFTWDRMIEEYIQVYQEILYRHH
Z blastp iščemo podobne proteine, ki so bolje anotirani. Z iskanjem po zbirki “non-redundant” najdemo podobne proteine, ki pripadajo glikoziltransferazni družini 4, zato lahko sklepamo, da je naš protein zagotovo član glikoziltransferazne družine 4. Po nizki E-vrednosti (0) in visoki podobnosti (do 80 %) podobnih proteinov sklepamo, da naš protein pripada rodu Syntrophomonas. Zadetek iskanja je na sliki 3.
Slika 3: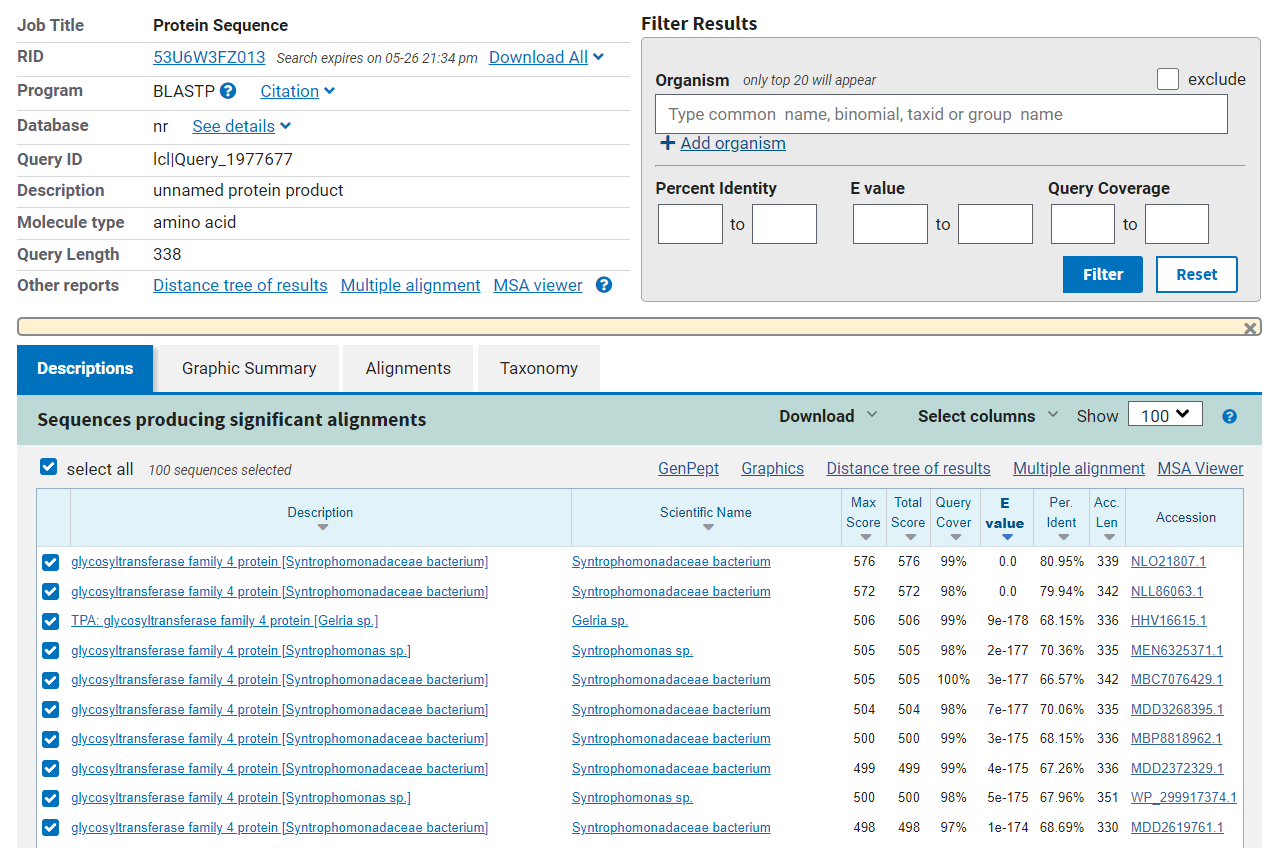
###Lokalizacija in topologija proteina Naš in našemu podobni proteini nimajo označene lokalizacije ali topologije, manj podobni proteini, ki so bolje anotirani, pa so premalo podobni, da bi lahko po podobnosti sklepali lokalizacijo in topologijo našega proteina. Za podatke o lokalizaciji se obrnemo na značilnosti glikoziltransferazne družine 4. Iz Interpro razberemo, da se proteini te družine nahajajo na membrani, ampak na podlagi primanjkljaja daljše regije hidrofobnih aminokislinskih ostankov in AlphaFold modela lahko sklepamo, da naš protein ni transmembranski, ampak le periferni membranski protein. Ob pogledu AlphaFold modela in glede na Pfam in NCBI CDD lahko topologijo našega proteina opišemo kot protein z dvema domenama, z visoko strukturno, a majhno sekvenčno homologijo. Vsaka domena vsebuje motiv Rossmanovega zvitja. Domeni sta ločeni s praznino, ki vsebuje vezavno mesto za substrat in omogoča fleksibilnost proteina.
###Velikost proteina Velikost lahko glede na naše aminokislinsko zaporedje izračunamo s pomočjo orodja ProtParam. Masa našega proteina je 38552 Daltona.
###Domenska zgradba Domensko zgradbo lahko določimo s pomočjo podobnih proteinov. Ker so proteini nazorneje označeni v UniProt, izvedemo Uniprot BLAST iskanje z našim aminokislinskim zaporedjem. Iskanje omejimo na zbirko UniProtKB in na rod “Syntrophomonas”. Iz rezultatov, vidnih na sliki 4, in lokalne poravnave proteinov z našim lahko določimo domensko zgradbo našega proteina in njeno ohranjenost. Z domensko anotacijo podobnega proteina A0A970PZH0 in na podlagi dobre lokalne poravnave z našim proteinom lahko sklepamo, da naš protein vsebuje dve domeni: glikoziltransferazno 4-like N-terminalno domeno in glikoziltransferazno 1 domeno, kar je značilno za glikoziltransferaze družine 4.
Slika 4: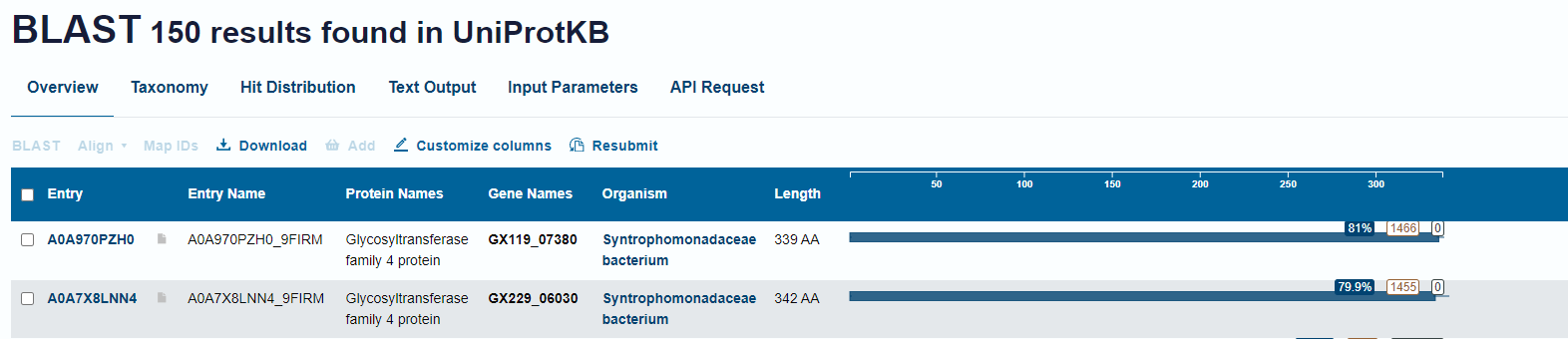
###Post-translacijske modifikacije Post-translacijskih modifikacij za naš protein ne moremo določiti, ker proteini, podobni našemu iz glikoziltransferazne družine 4, nimajo anotiranih post-translacijskih modifikacij. Proteini iz glikoziltransferazne družine 4, ki pa imajo anotirane post-translacijske modifikacije, izkazujejo premajhno podobnost z našim proteinom, da bi lahko na podlagi njihovih post-translacijskih modifikacij sklepali o post-translacijskih modifikacijah našega proteina.
###Sorodni proteini Filogenetsko drevo podobnih proteinov naredimo z iskanjem Uniprot BLAST po zbirki UniProtKB, omejenim na družino Syntrophomonadaceae. Zaporedja zadetkov s 700 točkami ali več shranimo v FASTA formatu in seznamu zaporedij dodamo še naše aminokislinsko zaporedje. Ta mejnik točk izberemo, saj je podobnost ostalih zadetkov znatno nižja, E-vrednost pa znatno višja. Seznam zaporedij v FASTA formatu nato uvozimo v Clustal Omega, kjer naredimo filogenetsko drevo. Filogenetsko drevo je prikazano na sliki 5.
Slika 5:
###Ohranjenost regij Iskanje z blastp po bazi “PDB” (rezultat na sliki 6) nam vrne eksperimentalno določeno strukturo podobnega prokariontskega proteina (7FG9). Lokalna poravnava (EMBOSS Water) nam pokaže, da sta si proteina podobna (slika 7). (7FG9) ima v Uniprot (Q8DIJ4) označeno vezavno mesto substrata (UDP), glikoziltransferazno 4-like N-končno domeno in glikoziltransferazno 1 domeno. Lokalna poravnava pokaže, da sta domeni dobro ohranjeni in da ima naš protein v aktivnem mestu zamenjan valinski aminokislinski ostanek za serinskega. Na podlagi te zamenjave sklepamo, da se katalitska aktivnost encima spremeni, čeprav je zamenjan le en izmed devetih aminokislinskih ostankov, ki gradijo vezavno mesto našega proteina. Valin je nepolaren in hidrofoben, medtem ko je serin polaren in hidrofilen, zaradi vezane hidroksilne skupine lahko tvori vodikove vezi. Zaradi zamenjave hidrofobnega ostanka s hidrofilnim se lahko spremeni lokalno okolje aktivnega mesta, zaradi česar je lahko ovirana vezava substrata ali stabilizacija prehodnega stanje. Hidroksilna skupina na serinu za razliko od valina se lahko povezuje z vodikovimi vezmi, kar lahko ali poveča ali zmanjša katalitsko aktivnost, odvisno od prvotne vloge valina v aktivnem mestu. Prav tako lahko sprememba velikosti in oblike stranske skupine zaradi zamenjava aminokislinskega ostanka povzroči konformacijsko spremembo aktivnega mesta, kar posredno vpliva na katalitsko aktivnost. Za natančnejšo določitev katalitske aktivnosti bi bilo potrebno izvesti ustrezne encimske teste.
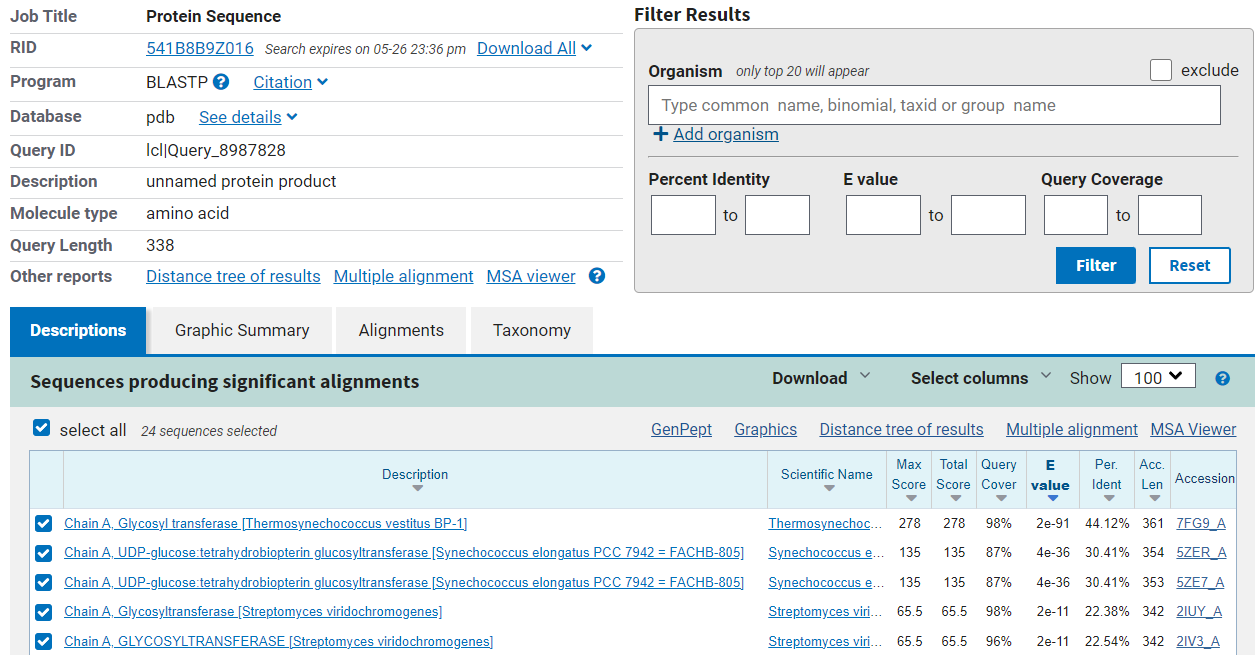
Slika 7: ###Eksperimentalne strukture podobnih evkariontskih proteinov
Iskanje poteka s pomočjo blastp po zbirki PDB z omejitvijo na evkariontske organizme. Rezultati so vidni na sliki 8. Ti proteini niso podobni našemu, zato imajo najverjetno drugačne značilnosti.
###Eksperimentalne strukture podobnih evkariontskih proteinov
Iskanje poteka s pomočjo blastp po zbirki PDB z omejitvijo na evkariontske organizme. Rezultati so vidni na sliki 8. Ti proteini niso podobni našemu, zato imajo najverjetno drugačne značilnosti.
Slika 8: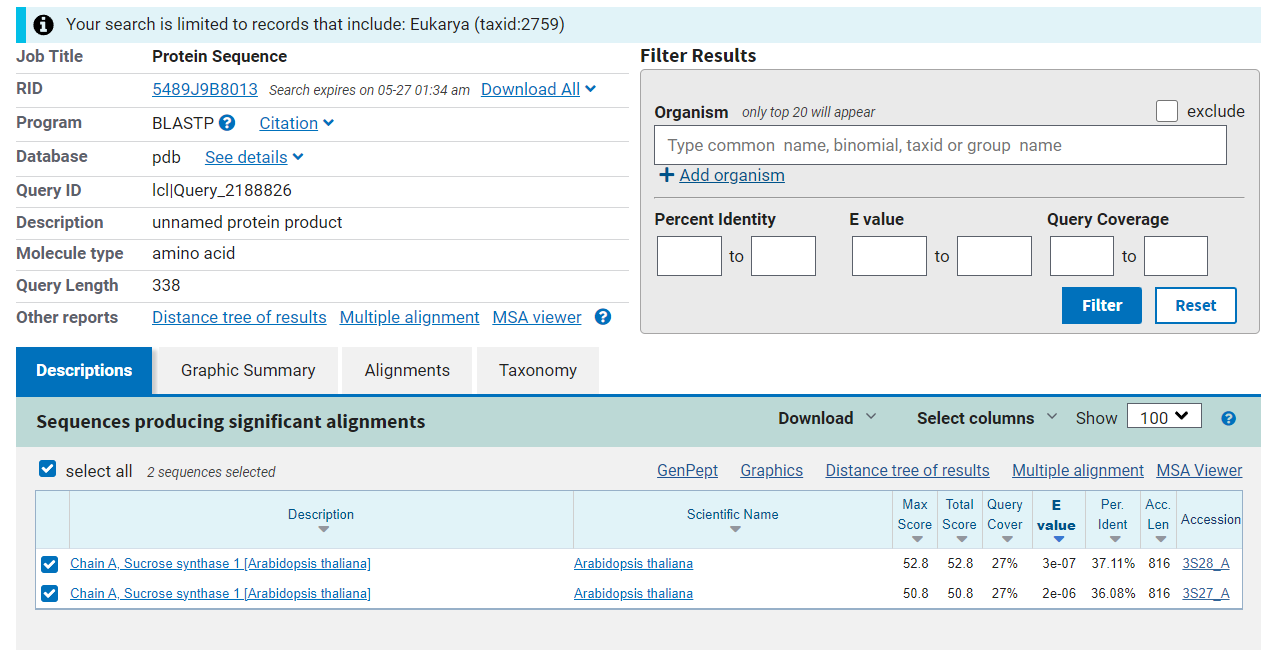
###Funkcionalna povezanost z drugimi proteini Na podlagi podobnosti s prokariontskim proteinom (7FG9), ki je homodimer in iz podobne glikoziltransferazne družine 4, ki so homo-n-meri, je možno, da je tudi naš protein homodimer ali homo-n-mer.
###Model strukture Model strukture našega proteina narejen z AlphaFold je viden na sliki 9. Surovi podatek iz AlphaFolda je viden na sliki 10. Sklepamo, da je model našega proteina zelo zanesljiv, saj je vrednost pLDDT za skoraj vse regije našega proteina med 90 % in 100 %, za preostale regije pa je med 70 % in 90 %. Glede na graf in nizko vrednost PAE je zanesljivost, da je večina regij na svojih relativnih mestih, visoka, zato sklepamo, da je model našega proteina zanesljiv.
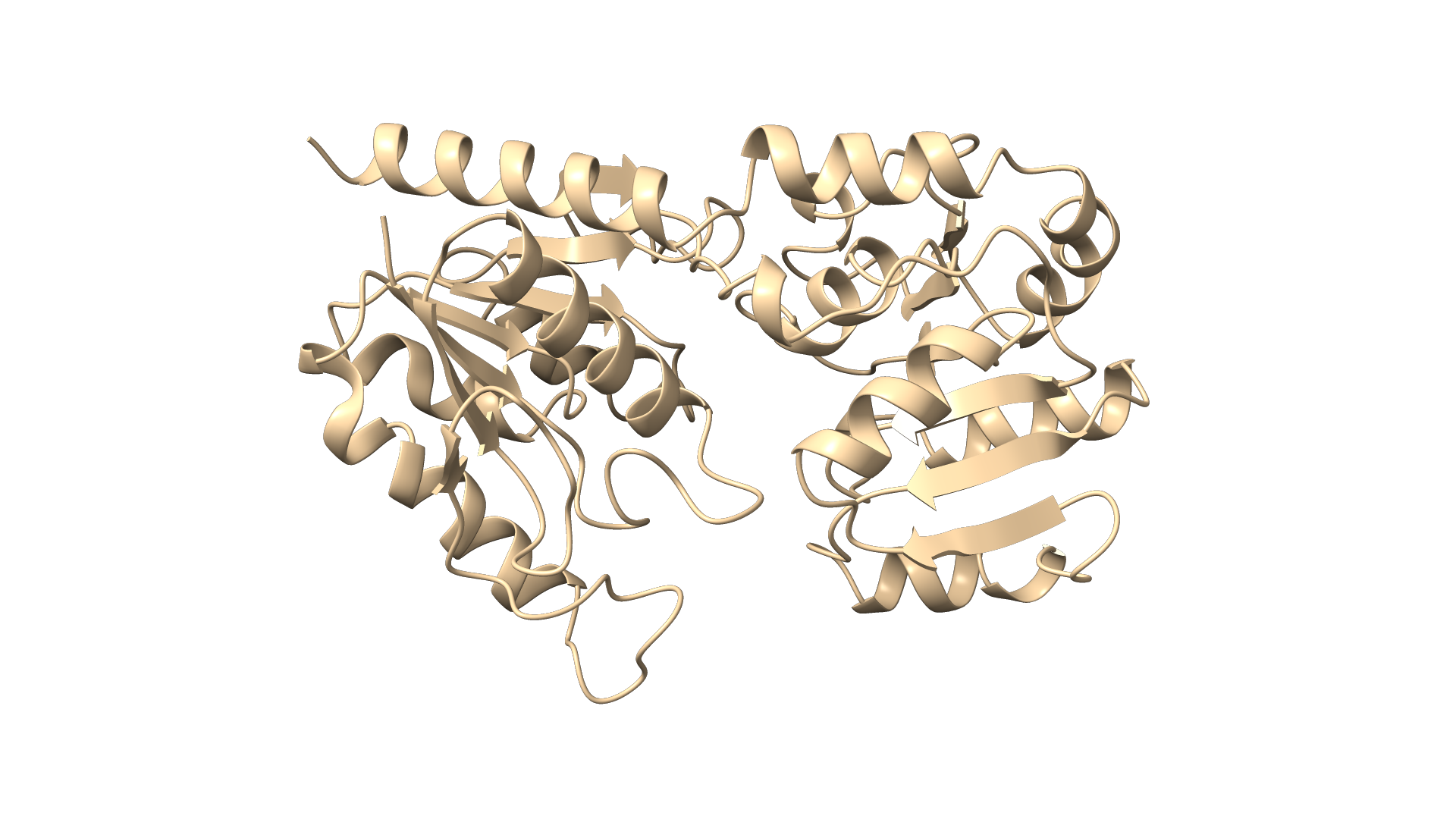
Slika 10: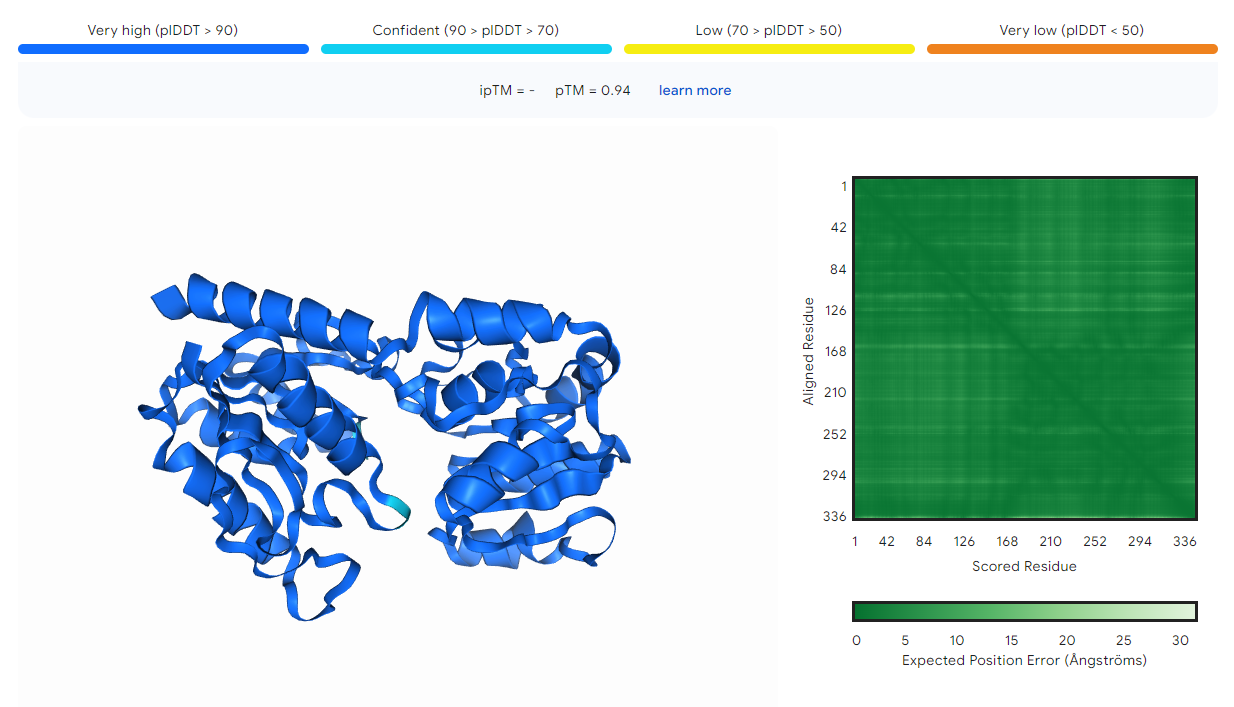
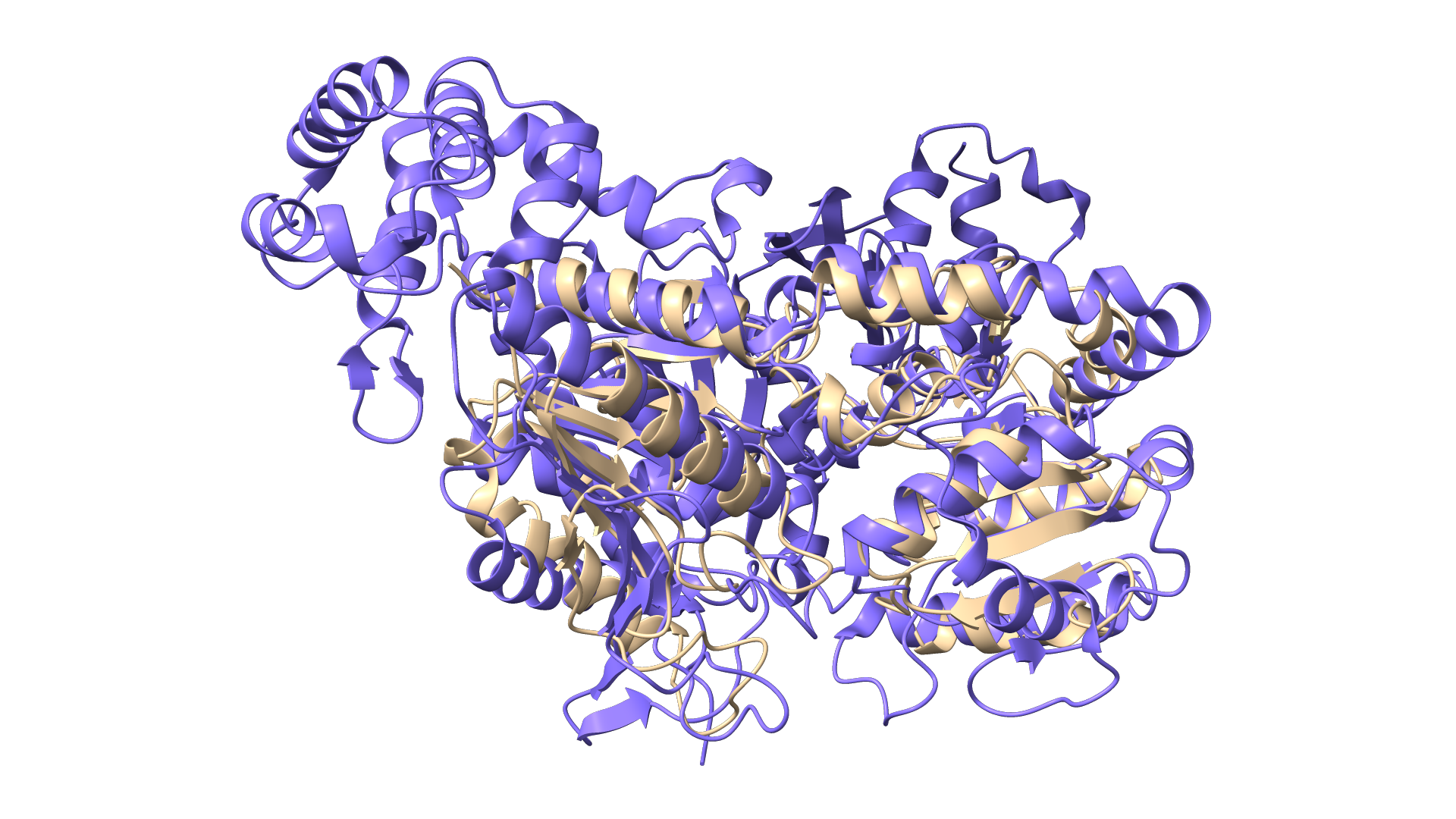
Zanesljivost modela potrjuje tudi ujemanje našega modela z modelom podobnega prokariontskega proteina (pridobljen eksperimentalno), ko izvedemo superpozicijo v programu ChimeraX. Superpozicija našega proteina in eksperimentalne strukture podobnega prokariontskega proteina je vidna na sliki 11.
Slika 11: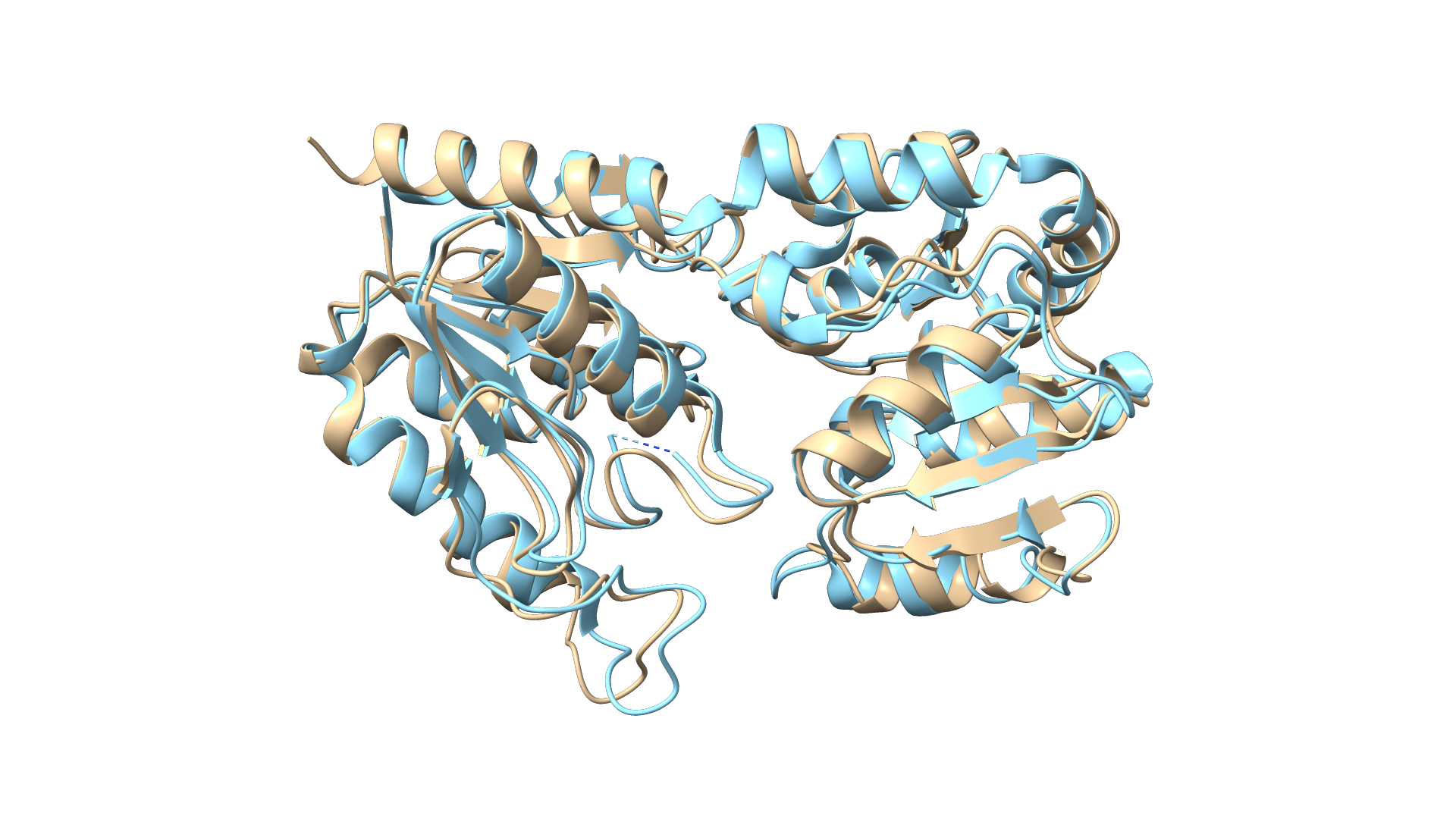
Ko izvedemo superpozicijo v programu ChimeraX, je ujemanje našega modela z modelom podobnega evkariontskega proteina (pridobljen eksperimentalno) slabše kot pri superpoziciji z modelom prokariontskega proteina, kar je posledica slabše podobnosti evkariontskega proteina našemu. Vseeno se modela na nekaterih mestih dobro ujemata, zato je naš model (predvsem na podlagi parametrov programa AlphaFold in ujemanja s prokariontskim proteinom) zanesljiv. Superpozicija našega proteina in eksperimentalne strukture podobnega evkariontskega proteina je vidna na sliki 12.
Slika 12: