VAJA: Modeliranje#
Pri modeliranju struktur proteinov lahko uporabljamo različne pristope. Nekaj let nazaj so dali najboljše rezultate v smislu pravilnosti (podobnosti eksperimentalni strukturi) pristopi na osnovi homologije, sploh če je bila med aminokislinskima zaporedjema tarče in znane eksperimentalno določene strukture visoka identičnost. Najnovejši pristopi, ki temeljijo na umetni inteligenci (UI; angl. artificial inteligence, AI), izračunajo danes izredno dobre modele struktur – med ta orodja spada AlphaFold.
Od orodij za modeliranje struktur proteinov se morda najpogosteje uporabljajo naslednja:
SWISS-MODEL za modeliranje na osnovi homologije, predvsem zaradi enostavnega vmesnika in hitrega izračuna modelov,
I-TASSER, ki je pred prihodom orodij na osnovi AI bilo eno izmed boljših orodij za modeliranje na osnovi homologije ter ab initio modeliranje krajših segmentov,
Rosetta, ki jo je treba z nekaj truda nainstalirati lokalno, dostopna pa je tudi preko strežnika Robetta, z AI podkrepljena varianta pa se imenuje RoseTTAFold in je dosegljiva kot opcija na strežniku Robetta, ter
AlphaFold2, ki ga lahko poganjamo lokalno (bolje: na računski gruči) ali pa izkoristimo računsko infrastrukturo Google Colab - ColabFold oz. AlphaFold3, ki ga poganjamo preko za to namenjenega strežnika (potrebujemo Google račun).
SWISS-MODEL#
Eno izmed uporabnikom zelo prijaznih in za uporabo preprostih orodij za modeliranje na osnovi homologije je SWISS-MODEL:
Interaktivno modeliranje zaženemo s pritiskom na gumb Start Modelling. Kot vhodni podatek vnesemo ak- zaporedje v formatu FASTA (ali kar UniProt kodo zapisa), eventuelno lahko “projektu” določimo ime in opcijsko vpišemo še e-poštni naslov (v primeru, da modeliranje traja dlje in nas o rezultatih obvesti prek e-pošte). Na voljo imamo iskanje po predlogah (Search For Templates), kjer nam predlaga eno ali več možnih struktur kot predloge (izberemo eno ali več, za vsako nam naredi ločen model), ali pa kar kliknemo Build Model, kjer program sam izbere zanj najbolj smiselno predlogo in izračuna en model.
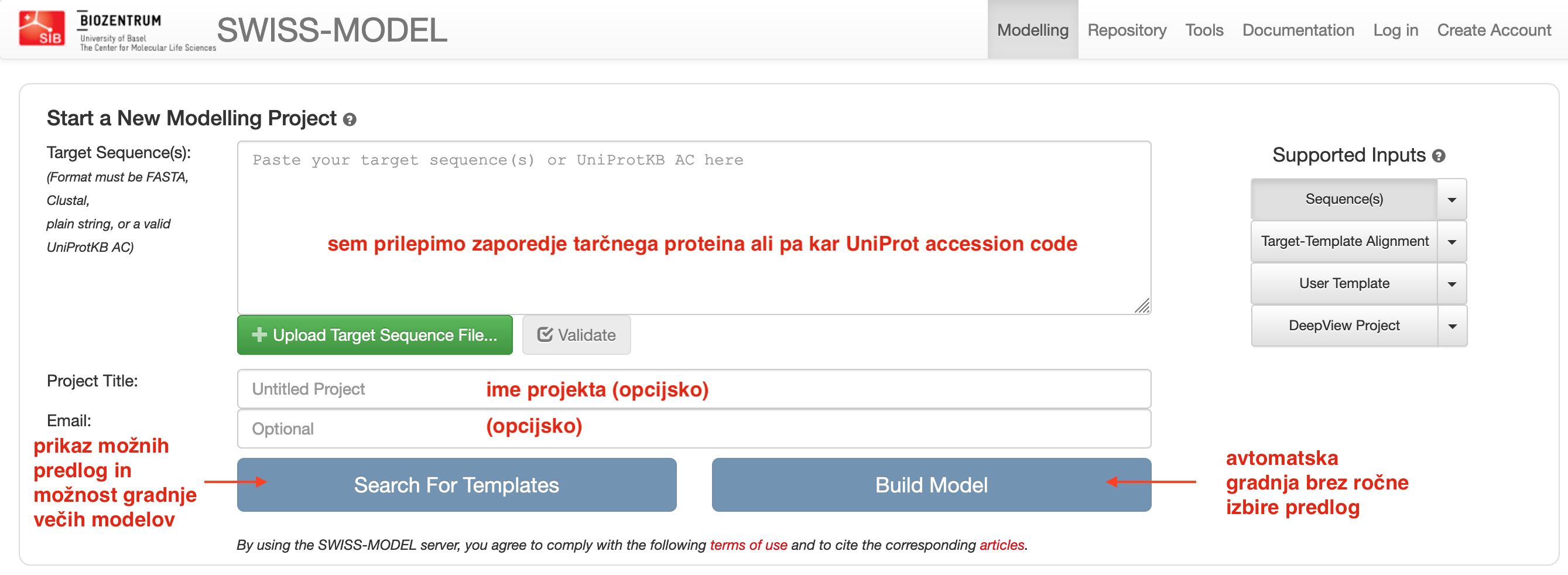
Podrobnosti in navodila so opisana v dokumentaciji. Posebej je potrebno omeniti parametra, izračunana tekom evaluacije modelov:
GMQE (Global Model Quality Estimate) je ocena kvalitete, ki poda na osnovi napovedi lDDT, in
QMEANDisCo global score, kjer gre za povprečje ocene kvalitete po posameznem aminokislinskem ostanku.
AlphaFold2#
Najlažji način za poganjanje AlphaFold2 je v obliki ColabFold iPython zvezka v storitvi Google Colab, kar je opisano v posebnem članku, zvezki za različne variante ColabFold pa so dostopni na repozitoriju ColabFold na GitHub. Osnovne variante, ki so na voljo, so:
AlphaFold2_mmseq2, ki za izračun poravnav uporablja MMseqs2 in HHsearch (hitreje kot Jackhammer),
AlphaFold_batch, ki prav tako uporablja MMseqs2, zvezek pa je prilagojen za pošiljanje več tarčnih zaporedij hkrati,
AlphaFold2, ki za izračun poravnav uporablja Jackhammer, gre pa za varianto od DeepMind (Google).
Navodila za ColabFold so dostopna na dnu iPython zvezka, uporaba pa je nazorno prikazana tudi na relativno dolgem videoposnetku na Youtube.
Na kratko, enostavno modeliranje monomernega proteina lahko z AlphaFold2_mmseq2 izvedemo tako:
V polje
query_sequencevnesemo aminokislinsko zaporedje.Kot
jobnamenastavimo poljubno ime (uporabimo ime proteina, pri dolžini in uporabljenih znakih je dobra razumnost.)Pri
num_relaxizberite, koliko končnih modelov želite energijsko minimizirati (relaksirati) z uporabo Amber. Za enostavne primere to kar pustite na 0, saj simulacija molekulske dinamike traja kar precej časa.Pri
template_modeizberite, ali bo AlphaFold2 poizkusil najti v zbirki PDB (dejansko gre za reducirano obliko pdb70, prag identičnosti 70 %) strukture s podobnim ak zaporedjem. To lahko tudi izključite (pri računu se upošteva samo stike med ak ostanki na osnovi analize poravnav), lahko pa naložite svojo predlogo (v formatu mmCIF).Ostale nastavitve so še glede MSA (katera zaporedja vzamemo in pa tvorba parov zaporedij, slednje je pomembno za modeliranje kompleksov pri čemer se združujejo zaporedja v pare le, če so vsa iskalna zaporedja (za vse verige kompleksa) prisotna v določeni taksonomski enoti).
Za modeliranje monomernega proteina pustimo
model_typena auto, prav tako število ciklov recikliranja.Priporočljivo je vključiti, da se rezultati na koncu zapakirajo v datoteko zip in naložijo v Google Drive (
save_to_google_drive).Zaženemo vse celice (najlažje preko Runtime > Run all).
Pozor:
Ne pretiravajte z velikostjo proteina, saj bo sicer na vam dodeljenem računskem viru na Google Colab zmanjkalo spomina in se bo modeliranje avtomatsko prekinilo.
Zavihka v brskalniku, kjer teče AlphaFold2, ne zapirajte, saj je pri brezplačnem računu obvezna uporaba interaktivnega načina. Lahko se zgodi, da se bo med modeliranjem pojavilo tudi pozivno okno, ki bo od vas zahtevalo interakcijo (preverjanje vaše prisotnosti).
Da bi bili modeli, izračunani z AlphaFold2, dostopni čim širši javnosti, so v EBI pripravili podatkovno zbirko vnaprej izračunanih modelov številnih proteinov, dne 2023-04-18 je šlo za modele več kot 200 milijonov proteinov. Med drugim gre za celoten človeški proteom ter proteome nekaterih modelnih organizmov, vsi modeli pa so dostopni preko posebne spletne strani AlphaFold DB, povezave do modelov pa so dodane tudi v kategorijo Structure v zbirki UniProt.
Modele si lahko na Strani UniProt oz. v podatkovni zbirki na EBI ogledamo kot 3D modele, pobarvane glede na zanesljivost napovedi strukture (pLDDT, per-residue confidence score, v razponu od 0 do 100). Pri regijah z nizko pLDDT gre pogosto za strukturno neurejene regije.
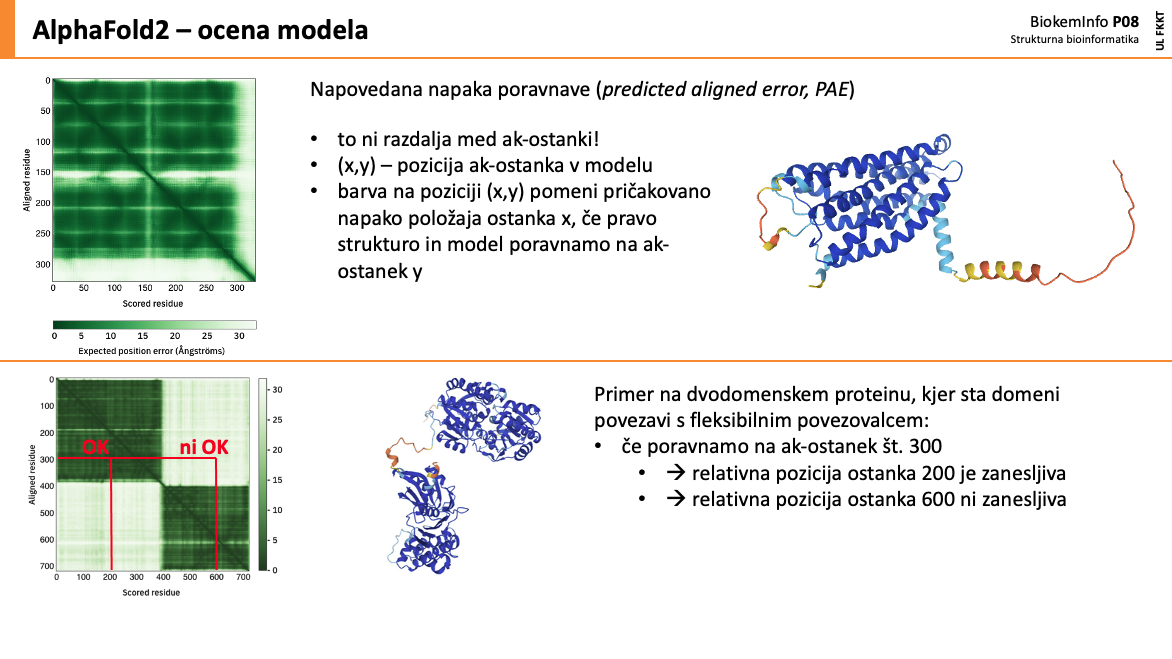
Pri analizi modelov ne pozabite na pLDDT in PAE:

AlphaFold3#
AlphaFold3 lahko najenostavneje poganjamo preko strežnika z izredno prijaznim uporabniškim vmesnikom – AlphaFold Server. Na voljo je modeliranje posameznih proteinov, posameznih nukleinskih kislin ter kompleksov, zna pa tudi dodati ligande in ione in nekatere post-translacijske modifikacije. Modeli so načeloma boljši kot pri AlpaFold2, še posebej v primerih, kjer gre za proteine, za katere ni moč najti veliko podobnih zaporedij, t.i. proteine “sirote” (angl. orphans).
Vmesnik je resnično enostaven in ga verjetno ni potrebno posebej razlagati, je pa na voljo spletni vodič.
Naloga#
Vsak si naj izbere en protein dolžine največ 300 aminokislinskih ostankov (priporočam, da izberete bolj “eksotične” proteine ali pa proteine iz “eksotičnih” organizmov), za katerega:
Z iskanjem po zbirki PDB preko programa BLAST preverite, ali je znana struktura točno tega proteina; če je znana struktura, ki pokriva večji del zaporedja izbranega proteina, si izberite drug protein.
Model proteina izračunajte z uporabo AlphaFold2_mmseq2 po navodilih zgoraj ali AlphaFold3.
Izdelajte tudi model s pomočjo programa SWISS-MODEL, in sicer za celotni protein (seveda odstranite morebitni signalni peptid in/ali proregije).
Izračunane modele in pa strukturo, uporabljeno kot predlogo v SWISS-MODEL, naložite v program UCSF Chimera (ali ChimeraX) in primerjajte preko superpozicije (MatchMaker). Lahko uporabite tudi MolStar.
Odgovorite na vprašanji:
Ali model iz SWISS-MODEL pokriva celotno ak zaporedje, ki ste ga uporabili? Zakaj da oz. ne?
Kako je z zanesljivostjo modela vzdolž aminokislinskega zaporedja (tako za SWISS-MODEL kot AlphaFold2/3)? Je to kakorkoli povezano s pričakovano strukturo (ali strukturno neurejenostjo) določenih regij zaporedja (npr. zanke)?
Ocenite biološko smiselnost modela glede na značilnosti zaporedja, anotirane v Uniprot, oz. glede na značilnosti dobro anotiranega homolognega proteina podobne dolžine.