M-coffee, poravnava zaporedij#
Avtorja: Jan Kogovšek, Klara Razboršek
Namen vaje#
Spoznati se s spletnim orodjem M-Coffee. S pomočjo tega orodja poravnati različna zaporedja (aminokislinska in nukleotidna) in ugotoviti ustreznost poravnav.
Program#
Program: M-Coffee
Avtorji programa: Dr. Cedric Notredame, Center for Genomic Regulation (CRG)
Reference:
Notredame, C.; Higgins, D.; Heringa, J., T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. Journal of Molecular Biology 302, 205-217. 10.1006/jmbi.2000.4042
“Manual.” https://petrov.stanford.edu/software/src/T-COFFEE_distribution_Version_5.65/doc/t_coffee_tutorial.htm#_Toc148261707 (pridobljeno dne Mar. 26, 2022).
Opis programa#
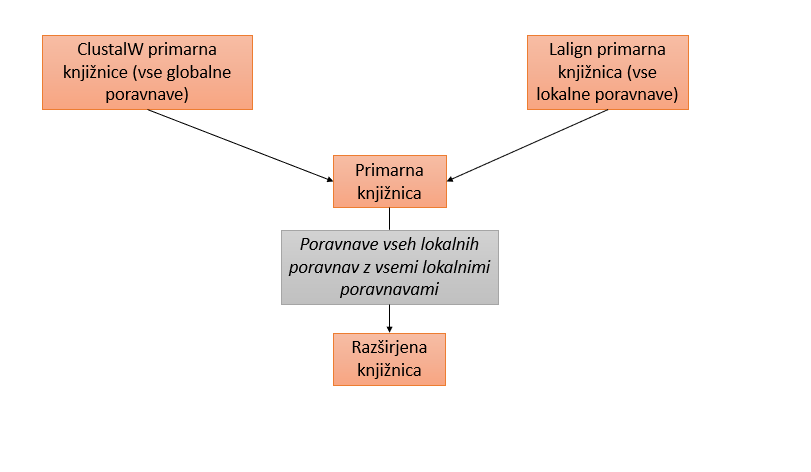
T-Coffee in njena pod-različica M-Coffee sta metodi za poravnavo več zaporedij. Algoritem temelji na ustvarjanju knjižnice poravnav, v kateri ima vsaka poravnava neko pripisano težo. Končna poravnava je sestavljena iz delčkov teh večih poravnav, ki daje največjo vsoto teže.
Proces ustvarjanja poravnave s T-Coffee se začne z generiranjem dveh knjižnic: s pomočjo ClustalW se ustvari knjižnica vseh možnih globalnih poravnav, s pomočjo Lalign pa knjižnica vseh lokalnih poravnav. Vsaki izmed teh poravnav se pripiše njena teža. Nato se ustvari tako-imenovana razširjena knjižnica, v kateri poravnamo vse globalne poravnave z vsemi lokalnimi in obratno. Šele ko je program ustvaril to razširjeno knjižnico začne ustvarjati optimalno poravnavo našega zaporedja, ki bo imel, glede na vsoto posameznih delov, največjo težo.
M-Coffee združuje več metod, ki ustvarjajo skupno knjižnico in s tem ustvarijo boljše poravnave. Uporablja se tako za poravnave nukleotidnih, kot tudi aminokislinskih zaporedij.
Vhodni podatki#
Vhodni podatki so lahko aminokislinska ali nukleotidna zaporedja, pomembno je le, da so ta v FASTA formatu. Lahko jih vpišemo ločeno v program, lahko pa jih vpišemo v beležnico (windows) oz. texteditor (macOS) ter datoteko naložimo v program.
Navodila#
Vhodni podatki#
Kot vhodne podatke uporabite:
aminokislinsko zaporedje človeške ligaze 1 (human ligase 1, UniProt ID P18858)
aminokislinsko zaporedje mišje ligaze 1 (mouse ligase 1, UniProt ID P37913)
aminokislinsko zaporedje glikoproteina gp160 virusa HIV 1 (HIV 1, UniProt ID P04578)
aminokislinsko zaporedje glikoproteina gp160 virusa HIV 2 (HIV 2, UniProt ID P15831)
Postopek dela#
Odpremo M-Coffee
Na UniProt-u poiščemo aminokislinsko zaporedje proteinov podanih v vhodnih podatkih
Zaporedje parov lahko prekopiramo direktno v M-Coffee ali pa ga shranimo na računalnik v beležnico (windows) oz. texteditor (macOS) ter to datoteko naložimo v program.
Stisnemo submit in počakamo na reyultate.
Poženemo tudi poravnavo v Uniprotu-u, kjer poravnavo izvede Clustal Omega.
Enak postopek ponovimo tudi za naslednji par.
Pričakovani rezultati in razlaga#
Zaslonski sliki prikazujeta rezultat poravnave v programu M-Coffee, kjer vidimo visoko ujemanje zaporedij, kar nakazuje na homolognost proteinov.
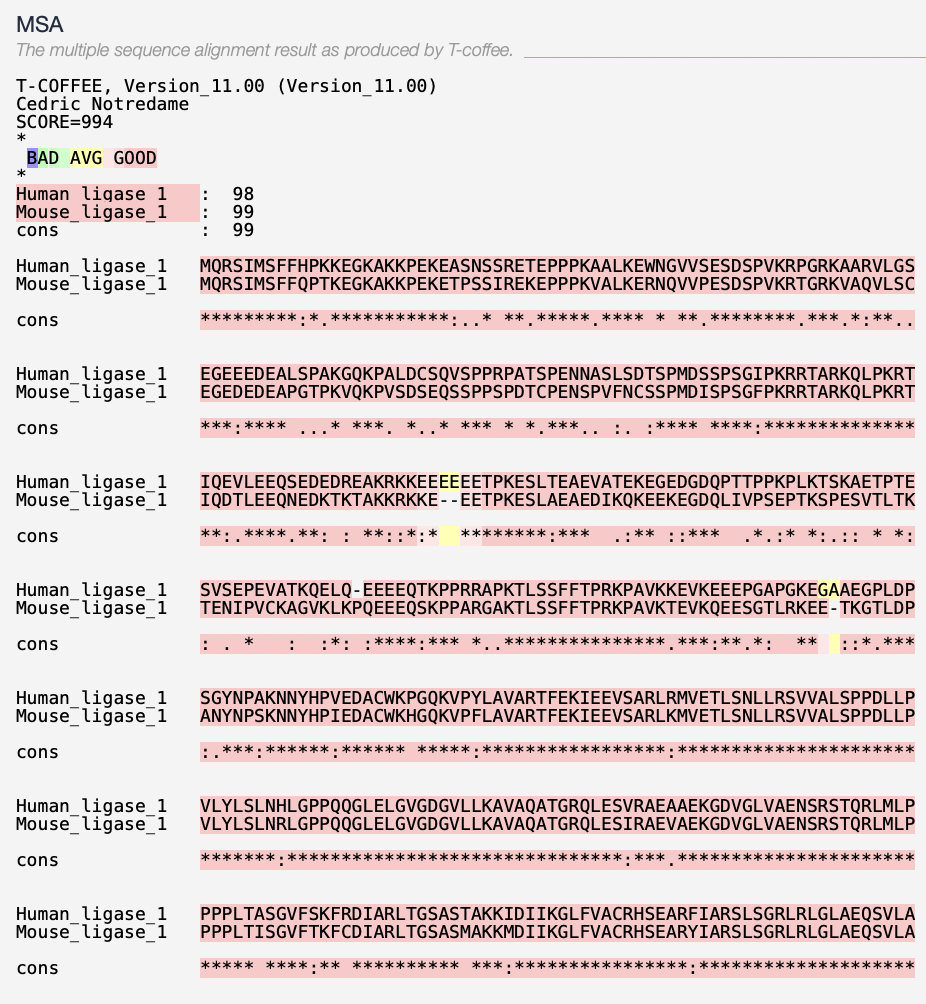

Slika prikazuje poravnavo zaporedja s Clustal Omega, kjer opazimo, da se dobro ujema s poravnavo v M-Coffee, zato sklepamo, da je program natančno poravnal aminokislinsko zaporedje.

Pri primerjavi glikoproteina gp160 virusne ovojnice virusa HIV 1 in 2 opazimo, da se ne ujemata v celoti, hkrati pa po primerjavi s Clustal Omega poravnavo opazimo, da je celotna poravnava drugačna, saj M-Coffee poravna na začetku brez vrzeli, medtem ko Clustal Omega na začetku pri HIV 2 vstavi vrzeli.

Slika je poravnava v programu Clustal Omega

Iz rezultatov lahko sklepamo, da M-Coffee bolje poravna homologna zaporedja, kot zaporedja, kjer so si vrste bolj oddaljene (primer HIV 1 in HIV 2). To se tudi sklada s komentarjem razvijalcev programa, ki so sami napovedali, da je orodje bolj primerno za primerjavo homolognih zaporedij.